[1]:
import os
output_dir = "output/batch_benchmark"
os.makedirs(output_dir, exist_ok=True)
Batch Effect Benchmark
HVG
[2]:
# Initialize
source_path = "../REVISE/results"
task = "spot"
patient_id = "P2CRC"
result_path = f"{source_path}/{task}/{patient_id}"
data_path = f"../REVISE/data/{task}/{patient_id}"
parts = ["part3", "part1", "part2"]
batch_nums = [1, 2, 3, 4]
metrics = ["PCC", "SSIM", "MSE"]
spot_sizes = [50, 100, 150, 200]
gene_type = "HVG"
gene_num = 50
[ ]:
import pandas as pd
import scanpy as sc
from tqdm import tqdm
import matplotlib.pyplot as plt
import seaborn as sns
def get_genes(result_path, data_path, part, spot_size, batch_num, gene_type = "HVG", gene_num = 50, test_genes = None):
## Get HVG or HEG or ALL
spot_path = os.path.join(data_path, f"cut_{part}", f"spot_{spot_size}")
save_path = os.path.join(result_path, part, f"{spot_size}_{batch_num}", "select_gene")
os.makedirs(save_path, exist_ok=True)
gene_file = f"{save_path}/{gene_type}_{batch_num}_genes_{gene_num}.txt"
if os.path.exists(gene_file):
# print(f"Find {gene_type} genes in {gene_file}")
with open(gene_file, "r") as f:
genes = f.read().splitlines()
return genes
st_path = f"{spot_path}/xenium_spot.h5ad"
adata = sc.read(st_path)
if test_genes is not None:
overlap_genes = [gene for gene in test_genes if gene in adata.var_names]
adata = adata[:, overlap_genes]
if gene_type == "HVG":
sc.pp.filter_genes(adata, min_cells=1)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.highly_variable_genes(adata, flavor='seurat_v3', n_top_genes=gene_num)
genes = adata.var[adata.var['highly_variable']].index.tolist()
elif gene_type == "HEG":
adata.var['sum'] = adata.X.toarray().sum(axis=0)
adata.var.sort_values('sum', ascending=True, inplace=True)
genes = adata.var.head(gene_num).index.tolist()
else:
genes = adata.var_names
with open(gene_file, "w") as f:
f.write("\n".join(genes))
# print(f"Save {gene_type} genes to {gene_file}")
return genes
def get_merge_df(result_path, data_path, part, metric, spot_sizes, batch_nums):
merge_df = pd.DataFrame()
for spot_size in tqdm(spot_sizes, desc="spot_sizes"):
for batch_num in batch_nums:
metric_file = f"{result_path}/{part}/{spot_size}_{batch_num}/metrics_normalized.csv"
df = pd.read_csv(metric_file, index_col=0)
genes = get_genes(result_path, data_path, part, spot_size, batch_num, gene_type = gene_type, gene_num = gene_num, test_genes = df.index)
df = df.loc[genes]
df = pd.DataFrame({
'Method': batch_num,
'Value': df[metric].values,
'Spot_size': spot_size,
'Part': part,
'Metric': metric,
})
merge_df = pd.concat([merge_df, df])
merge_df.reset_index(drop=True, inplace=True)
return merge_df
def plot_comp_seg(merge_df, metric, part, ax):
spot_size_order = [50, 100, 150, 200]
method_order = [1,2,3,4]
custom_palette = {
1: '#80a9c8',
2: '#e89786',
3: '#8ccfd9',
4: '#b39a94',
}
# boxplot
sns.boxplot(
data=merge_df,
x='Spot_size',
y='Value',
hue='Method',
order=spot_size_order,
hue_order=method_order,
palette=[custom_palette[m] for m in method_order],
width=0.6,
fliersize=2,
showfliers=False,
ax=ax
)
# ax.set_title(f'{part}', fontsize=11, pad=8)
# ax.set_xlabel('Spot Size', fontsize=10)
ax.set_ylabel(metric, fontsize=10)
ax.xaxis.set_visible(False)
if metric == "PCC":
ax.set_ylim(0, 1.02)
elif metric == "SSIM":
ax.set_ylim(0.5, 1.02)
elif metric == "MSE":
ax.set_ylim(1e-5, 1e-2)
ax.set_yscale('log')
if ax.get_legend() is not None:
ax.legend_.remove()
ax.set_aspect('auto')
[ ]:
# compute mean
parts = ["part3", "part1", "part2"]
metrics = ["PCC", "SSIM", "MSE"]
spot_sizes = [50, 100, 150, 200]
save_dir = f"{output_dir}/{gene_type}_mean"
os.makedirs(save_dir, exist_ok=True)
for part in tqdm(parts, desc="parts"):
for spot_size in tqdm(spot_sizes, desc="spot_sizes"):
merge_df = pd.DataFrame()
for batch_num in batch_nums:
metric_file = f"{result_path}/{part}/{spot_size}_{batch_num}/metrics_normalized.csv"
df = pd.read_csv(metric_file, index_col=0)
genes = get_genes(result_path, data_path, part, spot_size, batch_num, gene_type = gene_type, gene_num = gene_num, test_genes = df.index)
df = df.loc[genes]
df = df[metrics].mean(axis=0)
merge_df = pd.concat([merge_df, df], axis=1)
merge_df.reset_index(drop=True, inplace=True)
merge_df.index = metrics
merge_df.columns = ["Batch_1", "Batch_2", "Batch_3", "Batch_4"]
merge_df.T.to_csv(f"{save_dir}/{part}_{spot_size}_{gene_type}_{gene_num}.csv")
spot_sizes: 100%|██████████| 4/4 [00:01<00:00, 3.70it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 36.14it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 18.12it/s]
parts: 100%|██████████| 3/3 [00:01<00:00, 2.11it/s]
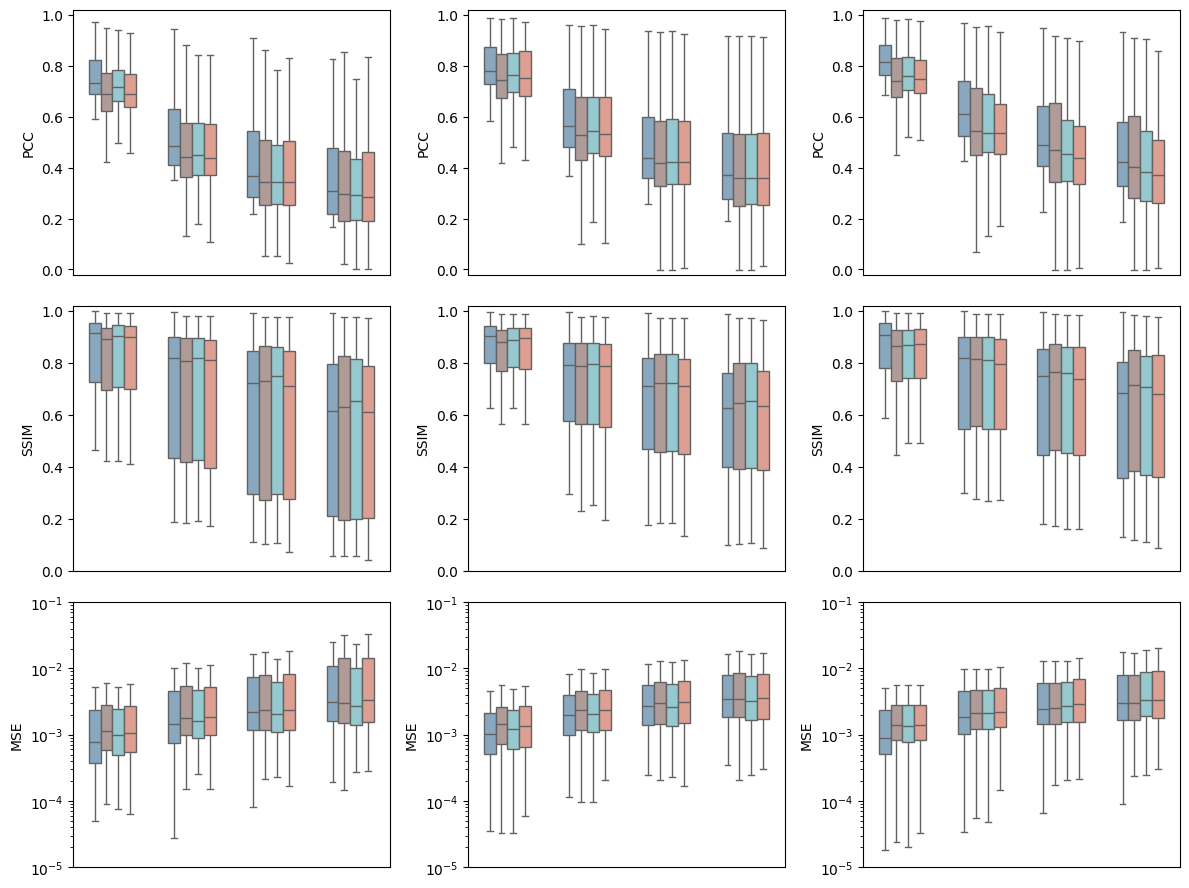
[9]:
fig, axes = plt.subplots(3, 3, figsize=(12, 9))
for i, part in enumerate(parts):
for j, metric in enumerate(metrics):
merge_df = get_merge_df(result_path, data_path, part, metric, spot_sizes, batch_nums)
ax = axes[j, i]
plot_comp_seg(merge_df, metric, part, ax)
plt.tight_layout()
plt.savefig(f"{output_dir}/batch_effect_{gene_type}_{gene_num}.pdf", dpi=300)
plt.show()
spot_sizes: 0%| | 0/4 [00:00<?, ?it/s]spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 160.35it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 166.75it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 164.43it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 147.27it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 166.60it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 166.84it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 164.62it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 166.86it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 166.11it/s]
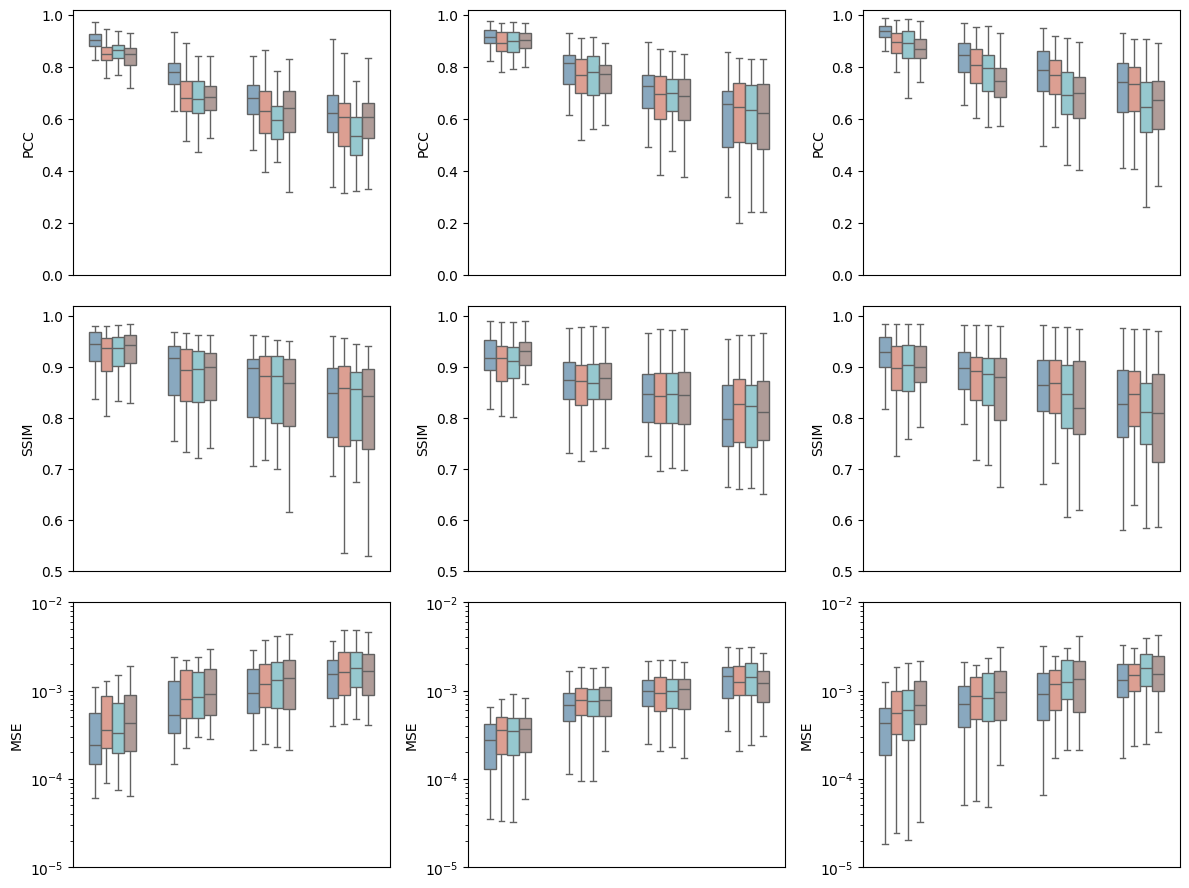
All genes
[33]:
gene_type = "ALL"
def plot_comp_seg(merge_df, metric, part, ax):
spot_size_order = [50, 100, 150, 200]
method_order = [1,2,3,4]
custom_palette = {
1: '#80a9c8',
2: '#b39a94',
3: '#8ccfd9',
4: '#e89786',
}
# Plot boxplots
sns.boxplot(
data=merge_df,
x='Spot_size',
y='Value',
hue='Method',
order=spot_size_order,
hue_order=method_order,
palette=[custom_palette[m] for m in method_order],
width=0.6,
fliersize=2,
showfliers=False,
ax=ax
)
# ax.set_title(f'{part}', fontsize=11, pad=8)
# ax.set_xlabel('Spot Size', fontsize=10)
ax.set_ylabel(metric, fontsize=10)
ax.xaxis.set_visible(False)
if metric == "PCC":
ax.set_ylim(-0.02, 1.02)
elif metric == "SSIM":
ax.set_ylim(0, 1.02)
elif metric == "MSE":
ax.set_ylim(1e-5, 1e-1)
ax.set_yscale('log')
if ax.get_legend() is not None:
ax.legend_.remove()
ax.set_aspect('auto')
[ ]:
fig, axes = plt.subplots(3, 3, figsize=(12, 9))
for i, part in enumerate(parts):
for j, metric in enumerate(metrics):
merge_df = get_merge_df(result_path, data_path, part, metric, spot_sizes, batch_nums)
ax = axes[j, i]
plot_comp_seg(merge_df, metric, part, ax)
plt.tight_layout()
plt.savefig(f"{output_dir}/batch_effect_{gene_type}_{gene_num}.pdf", dpi=300)
plt.show()
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 77.87it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 153.81it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 145.06it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 149.87it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 150.76it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 149.96it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 151.82it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 152.39it/s]
spot_sizes: 100%|██████████| 4/4 [00:00<00:00, 151.33it/s]